An uncommon genetic defect causes the most common disorder of lipid metabolism involving skeletal muscle. Fatty acids are a major source of fuel for human cells. They must be oxidized within mitochondria to release their energy, but some fatty acids cannot enter the mitochondria without first being joined to carnitine, an amino acid-like molecule.
Carnitine palmitoyltransferase, an enzyme that is embedded in the mitochondrial membrane, facilitates the transfer of long-chain fatty acids to the mitochondrial interior where they can be metabolized. Genetic deficiencies of carnitine palmitoyltransferase can cause a constellation of symptoms, get more info.
Carnitine Recycling Drives Long-Chain Fatty Acid Metabolism
Two forms of carnitine palmitoyltransferase, or CPT, reside within the two-layered mitochondrial membrane. The first, CPT1, is located on the outer membrane, and it is responsible for hooking a long-chain fatty acid to a molecule of carnitine. The carnitine-fatty acid complex is then transported into the mitochondria, where the second form of carnitine palmitoyltransferase, called CPT2, detaches the carnitine from the fatty acid.
The fatty acid is then metabolized, while the carnitine diffuses out of the mitochondrion to the cellular cytoplasm where it can be attached to another fatty acid and repeat the process.
CPT1 Deficiency
Three distinct isoforms of CPT1 have been identified in liver, muscle and brain. According to a 2014 article in “Molecular Aspects of Medicine,” the only known CPT1 deficiency involves the liver isoform, and it is exceptionally rare. People with CPT1 deficiency experience recurrent episodes of muscle weakness, confusion and low blood sugar, and they often develop enlarged livers.
Episodes are often initiated by fasting, acute illnesses or other situations that demand increased use of fatty acids for energy. Because fatty acids are not being efficiently utilized, they can build up in various tissues and cause organ damage. People affected by CPT1 deficiency are at risk for nervous system damage, seizures, liver failure, coma and sudden death.
CPT2 Deficiency
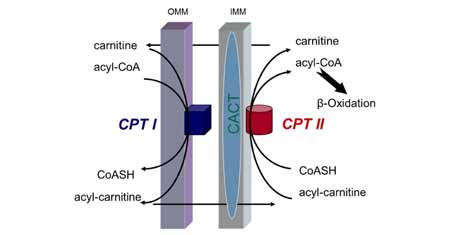
Defects in the gene that codes for production of CPT2 are much more common than those that impair CPT1 function. CPT2 deficiency manifests in at least three different forms. The lethal infantile form is uniformly fatal within the first few months of life. It usually presents within days of birth and is characterized by birth defects, liver and heart failure, abnormal cardiac rhythms, seizures and coma. The severe infantile form, which is the second type of CPT2 deficiency, typically presents within the first year of life and causes liver and heart failure, muscle weakness, seizures, headaches and abdominal pain. Sudden death may occur as the result of heart arrhythmias.
The adult “myopathic” form of CPT2 deficiency is the most common type. Symptoms can begin before puberty, but they may not begin until the six or seventh decade of life. Affected individuals experience intermittent attacks of weakness, muscle pain and myoglobinuria, which is brown-colored urine that results from the products of muscle damage appearing in the urine. Episodes are usually precipitated by prolonged exercise, cold exposure, fever or stress, but they can also be triggered by general anesthesia or sleep deprivation.
Treatment of Carnitine Palmitoyltransferase Deficiency
Some forms of CPT deficiency, such as the lethal infantile form, do not respond to treatment. Other forms can be partially ameliorated by restricting long-chain fatty acids in the diet, limiting total fat consumption to less than 20 percent of daily caloric intake, avoidance of fasting or prolonged exercise, supplementing with carnitine to help override the enzyme deficiency, increasing carbohydrate consumption and supplementing with medium-chain fatty acids to provide fuel molecules that don’t require CPT for transport.
According to Dr. Thomas Wieser, a neurologist at the Medical University of Vienna, Austria, people who suffer from CPT2 deficiency should avoid ibuprofen, general anesthesia, valproic acid and diazepam. Wieser also recommends genetic testing and counseling for affected individuals.